

Opis
Czy to badanie jest dla mnie?
Wykonaj badanie, jeśli….
- planujesz teraz bądź w przyszłości zajść w ciążę
- leczysz się wraz z partnerem z powodu niepłodności o niewyjaśnionej przyczynie
- jesteś wraz z partnerem objęta opieką i leczeniem w klinikach in-vitro, przygotowywane do zapłodnienia in vitro
- jesteś po licznych poronieniach samoistnych, które nie mają wyjaśnionego podłoża
Co da Ci to badanie?
Wykonując badanie CLARA sprawdzisz, czy jesteś nosicielem najczęstszych chorób genetycznych występujących w populacji polskiej. Tym samym dowiesz się czy istnieje ryzyko odziedziczenia tej choroby przez Twoje dziecko.
Badanie możesz wykonać przed podjęciem decyzji o macierzyństwie.
Czy wiesz, że każdy z nas jest nosicielem co najmniej 5-6 mutacji?
Mutacje to zmiany w genach, które nie zawsze muszą dawać objawy chorobowe. Dzieje się tak w przypadku nosicielstwa, czyli posiadania tylko jednej kopii zmienionego genu.
Sytuacja może ulec zmianie, jeśli zarówno Ty jak i Twój partner jesteście nosicielami tej samej mutacji w danym genie. Istniej wówczas 25% ryzyko, w przypadku części chorób genetycznych, że Wasze dziecko będzie chore. Ten genetyczny bagaż przekazujemy naszym dzieciom, niezależnie od wieku w jakim jesteśmy.
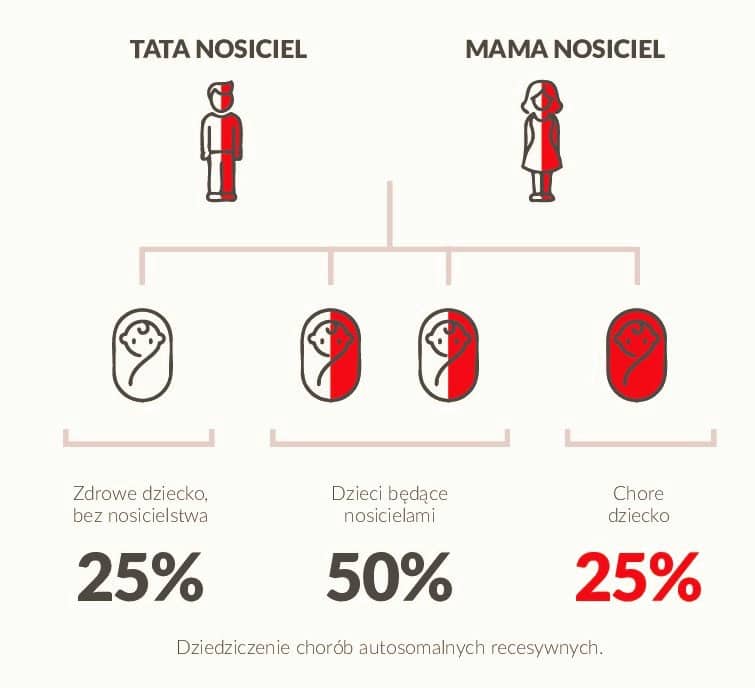
Jakie są rozwiązania, jeśli Ty i Twój partner jesteście nosicielami?
- Jeśli wynik potwierdzi nosicielstwo, macie możlwiość skorzystania z odpowiednich metod leczenia.
- Możecie świadomie wybrać inną formę zapłodnienia. Plan leczenia może obejmować – inseminację nasieniem dawcy, in vitro z dawstwem komórek jajowych lub przystąpienie do procedury in-vitro z dodatkową diagnostyką zarodka – (IVF + PGT-M – preimplantation genetic testing for monogenetic diseases)
- Dzięki tym rozwiązaniom, można zminimalizować ryzyko wystąpienia choroby u przyszłego dziecka, tej w kierunku której zostanie przpeprowadzona diagnostyka zarodka – PGT-M.
Genetyczne badanie zarodka – PGT-M
- Świadomie przygotujecie się na odpowiednie poprowadzenie ciąży (badania prenatalne potwierdzające lub wykluczające ryzyko wystąpienia choroby), ostatecznie zaplanowanie odpowiedniego leczenia chorego dziecka.
Co badamy w panelu?
Panel CLARA optima został stworzony z przede wszystkim z myślą o parach pochodzenia polskiego.
Obejmuje on wykrycie nosicielstwa najczęstszych chorób genetycznych, z uwzględnieniem najczęstszych mutacji w populacji słowiańskiej.
Istotne jest, aby panele nosicielstwa wykonała para, ponieważ ryzyko wystąpienia danej choroby u dziecka jest szacowane dla pracy planującej potomstwo.
| NOSICIELSTWO JEDNOSTKI CHOROBOWEJ |
GEN |
BADANE MUTACJE |
|---|---|---|
| Mukowiscydoza |
CFTR |
delF508, del2,3 |
| Rdzeniowy zanik mięśni (SMA) |
SMN1 |
delecja eksonu 7 |
| Zespół łamliwego chromosomu X |
FMR1 |
mutacja dynamiczna (CGG)n (badanie przesiewowe) |
| Niedosłuch wrodzony |
GJB2 |
c.35delG, c.35G>T, c.35G>A |
| Fenyloketonuria |
PAH |
c.1222C>t |
| Niedobór dehydrogenazy długołańcuchowych kwasów tłuszczowych (LCHADD) |
HADHA |
c.1528G>C |
| Niedobór dehydrogenazy acylo-CoA średniołańcuchowych kwasów tłuszczowych (MCADD) |
ACADM |
c.985A>G |
| Zespół Smitha-Lemliego-Opitza (SLOS) |
DHCR7 |
c.976G>T, c.452G>A, c.452G>C |
| Zespół Nijmegen |
NBN |
c.657_661del5 |
| Deficyt białka SCO2 (choroba mitochondrialna) |
SCO2 |
c.418G>A |
| Zespół Leigha (choroba mitochondrialna) |
SURF1 |
c.312_321del10insAT, c.845_846delCT |
Panele CLARA zostały stworzony na podstawie wiedzy klinicznej i laboratoryjnej, z wykorzystaniem doświadczeń różnych ośrodków genetyki medycznej w Polsce i są zgodne z rekomendacjami niżej wymienionych stowarzyszeń.
- Amerykańskie Towarzystwo Ginekologów i Położników zaleca, aby poinformować o badaniu nosicielstwa każdą kobietę planującą ciążę lub będącą w ciąży.
- Zgodnie z rekomendacjami ESHG (Europejskie Towarzystwo Genetyki Człowieka) oraz ACMG(Amerykańskie Kolegium Genetyki Medycznej i Genomiki) w skład panelu powinny wchodzić choroby o znanym obrazie klinicznym, a więc takie których znane są objawy chrakteryzujące samą chorobę oraz stopień jej zaawansowania. Wartość kliniczna paneli nosicielstwa jest istotniejsza niż liczba przebadanych genów.
| JEDNOSTKA CHOROBOWA |
CHARAKTERYSTYKA |
|---|---|
| Mukowiscydoza |
Najczęściej występująca na świecie choroba genetyczna skutkująca skróceniem życia. W przebiegu tej choroby dochodzi do zaburzenia funkcji gruczołów zewnątrz wydzielniczych, co prowadzi do niewydolności związanych z nimi narządów, przede wszystkim układów oddechowego oraz pokarmowego. |
| Rdzeniowy zanik mięśni (SMA) |
Druga najczęściej występująca choroba genetyczna skutkująca skróceniem życia. Polega na postępującym obumieraniu neuronów w rdzeniu kręgowym co prowadzi do osłabienia siły mięśni. Choroba może objawiać się w różnym wieku, jednak w około 70% przypadków pierwsze symptomy pojawiają się w okresie niemowlęcym lub wczesnym dzieciństwie. Obecnie SMA jest możliwe do wyleczenia dzięki zastosowaniu terapii genowej określanej mianem najdroższego leku świata. Jej koszt wynosi miliony złotych i nie jest objęta refundacją NFZ. |
| Zespół łamliwego chromosomu X |
Tzw. pełna mutacja (od 200 powtórzeń CGG) to jedna z najczęstszych przyczyn niepełnosprawności intelektualnej oraz objawów spektrum autyzmu uwarunkowanych genetycznie. Premutacja (zakres powtórzeń 55 – 199) wiąże się z ryzykiem przedwczesnego wygasania czynności jajników u kobiet (POI – ang. premature ovarian insufficiency). Ze względu na dynamiczny charakter mutacji nosicielstwo zwiększonej liczby powtórzeń trójnukleotydowych wiąże się z ryzykiem zwielokrotnienia do zakresu pełnej mutacji w kolejnym pokoleniu, co skutkuje wystąpieniem u dziecka zespołu łamliwego chromosomu X (FXS). |
| Niedosłuch wrodzony |
Niedosłuch jest jedną z najczęstszych chorób narządów zmysłów, natomiast gen GJB2 jest najważniejszym z dotychczas poznanych genów odpowiedzialnych za powstawanie niedosłuchu izolowanego. Mutacja c.35delG odpowiada za ok. 80% przypadków wrodzonego niedosłuchu spowodowanego mutacjami w genie GJB2. |
| Fenyloketonuria |
Chorobą metaboliczna prowadząca do gromadzenia się w organizmie fenyloalaniny. Z uwagi na defekt metabolizmu fenyloalaniny konieczne jest stosowanie diety eliminacyjnej niskofenyloalaninowej. W przypadku nieprzestrzegania diety bądź jej zbyt późnego wprowadzenia dochodzi do nieodwracalnych zmian neurorozwojowych takich min. upośledzenie rozwoju umysłowego oraz motorycznego. |
| Niedobór dehydrogenazy długołańcuchowych kwasów tłuszczowych (LCHADD) |
Wada genetyczna często określana jako „gen kaszubski” ze względu na częste występowanie mutacji w tej populacji. W przebiegu choroby dochodzi do nagłych spadków glukozy we krwi (np. po intensywnym wysiłku fizycznym, infekcji, której towarzyszą wymioty i biegunka lub podczas dłuższych przerw w jedzeniu). Gdy organizm nie może czerpać energii z posiłków zaczyna wykorzystywać własne kwasy tłuszczowe. Mutacja genu HADHA, odpowiedzialna za ich deficyt mu to jednak uniemożliwia. Niedobór LCHAD jest chorobą potencjalnie śmiertelną, jednak zdiagnozowany można leczyć za pomocą diety oraz unikania głodzenia. |
| Niedobór dehydrogenazy acylo-CoA średniołańcuchowych kwasów tłuszczowych (MCADD) |
Jest wrodzonym defektem mitochondrialnego utleniania kwasów tłuszczowych. Choroba ta charakteryzuje się szybko postępującym załamaniem metabolicznym, które często objawia się hipoglikemią hipoketotyczną, letargiem, wymiotami, drgawkami i śpiączką. Charakteryzuje się 25% ryzykiem zgonu przy pierwszym epizodzie. Podobnie jak w przypadku LCHAD należy unikać głodzenia. |
| Zespół Smitha-Lemliego-Opitza (SLOS) |
Mutacje w genie DHCR7 powodują brak lub zmniejszenie aktywności enzymu reduktazy 7-dehydrocholesterolu, co prowadzi do zahamowania szlaku endogennej syntezy cholesterolu w organizmie lub do zmniejszenia powstawania cholesterolu. Zespół ten charakteryzuje duża zmienność obrazu klinicznego, od postaci letalnych po łagodne. Objawem dominującym jest niepełnosprawność intelektualna, często w stopniu głębokim oraz zespół wad wrodzonych m.in. mózgu, serca, przewodu pokarmowego, nadnerczy, narządów płciowych zewnętrznych, a także cechy dysmorfii, m.in. małogłowie (mikrocefalia) i zrośnięcie palców (syndaktylia). Obecnie w Polsce, w stosunku do częstości nosicielstwa odnotowuje się zbyt małą liczbę rozpoznań choroby, co wiązane jest z niezdiagnozowaniem postaci łagodnych i atypowych, ale także prawdopodobnie wysoką letalnością w okresie płodowym oraz śmiertelnością w noworodkowym. Przypuszcza się, że SLOS jest jedną z najczęstszych chorób metabolicznych w populacji polskiej. |
| Zespół Nijmegen |
Choroba wywołana mutacjami w genie NBN kodującym nibrynę, białko biorące udział w mechanizmie naprawie pęknięć w dwuniciowym DNA zapewniając tym samym utrzymanie stabilności chromosomowej i integralności genomu. Na skutek mutacji w genie NBN dochodzi do zaburzeń naprawy DNA predysponujących do spontanicznych pęknięć chromosomów i ich rearanżacji. W konsekwencji choroba ta charakteryzuje się znaczną skłonnością do występowania nowotworów złośliwych, zwłaszcza glejaków i chłoniaków. Ponadto, choroba objawia się wewnątrzmacicznym opóźnieniem wzrastania płodu, a następnie zaburzeniem wzrastania, a także niedoborem odporności i cechami dysmorfii jak małogłowie czy mikrognacja. Większość dzieci umiera w pierwszej dekadzie życia z powodu nowotworów. Zespół ten występuje najczęściej w populacjach słowiańskich, co w 90% przypadków uwarunkowane jest założycielską mutacją c.657_661del5, określaną jako “mutacja słowiańska”, zwiększoną predyspozycję do powstawania nowotworów obserwuje się także u heterozygotycznych nosicieli mutacji. |
| Deficyt białka SCO2 (choroba mitochondrialna) |
Choroby mitochondrialne to schorzenia wynikające z zaburzeń w funkcjonowaniu i strukturze mitochondriów spowodowanych mutacjami w genomie mitochondrialnym lub jądrowym DNA kodującym białka specyficzne dla mitochondriów i regulujące ich działanie. Jednymi z częściej występujących defektów genetycznych są mutacje w genach SURF1 (zespół Leigha) i SCO2 (deficyt białka SCO2). Objawy chorób mitochondrialnych u dzieci są niespecyficzne i obejmują zestaw postępujących zmian w mózgu, mięśniach, gruczołach wydzielania wewnętrznego, narządach zmysłów (głuchota, oftalmoplegia) i innych tkankach i narządach. Co związane jest z faktem, iż mitochondria są organellami dostarczającymi komórkom energię niezbędną do ich prawidłowego funkcjonowania. |
| Zespół Leigha (choroba mitochondrialna) |
Bezboleśnie
Szybko i łatwo
Bez wychodzenia z domu
Badanie dostarczone do Ciebie w ciągu 24h.
Wynik badania w ciągu 14 dni.
Badania wykonywane jest z DNA pochodzącego ze śliny.
Jak to działa?

CZY ŚLINA JEST DOBRYM MATERIAŁEM?
Dowiedz się dlaczego badanie genetyczne ze śliny jest tak samo wiarygodne jak badanie z krwi – BADANIE ZE ŚLINY – WSZYSTKO CO POWINIENEŚ WIEDZIEĆ.
Opinie
Na razie nie ma opinii o produkcie.